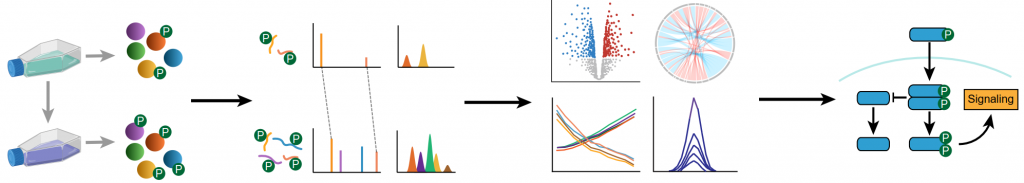
Proteoforms are the various molecular forms a protein can take, and they depend on several determinants (1). These include protein PTMs, which are among the essential modulators of protein functions. Theoretically, DIA-MS contains all the existing PTMs of a protein (2) and thus outperforms DDA-MS in identifying low-level or understudied PTMs. Another advantage of DIA-MS is its capability of discriminating isobaric PTMs located on different AAs from the same sequence (the so-called “site-specific issue”) (3).
Sample preparation is not different for PTM-DIA-MS compare to PTM-DDA-MS, except that offline fractionation is usually not required. The main hurdle of PTM-DIA studies lies in its data analysis strategies. The manual inspection of the proteome data for potential PTMs was initially used in histone (4, 5) and phosphorylation (6) studies. In these cases, the DIA-MS raw data were first searched in a spectrum-centric manner to generate un-modified (stripped) protein lists. A list of their modified sequences, with canonical histone PTMs, was subsequently generated in Skyline for targeted selection and quantitation. Multiplex Adduct Peptide Profiling (7), an alternative spectrum-centric approach, extracts the MS2 signals of interest from the DIA-MS spectra, overlays them with the coeluting MS1 signals, and thereafter performs signal trace verification. Keller et al. automated the signal extraction process and designed a scoring system to validate the results (8). Bekker-Jensen et al. applied predicted library to use for site-specific phosphorylation identification (9). Over 20,000 phosphopeptides could be quantified within a 15-min single-shot DIA-MS measurement, proving this strategy’s great potential. However, due to the highly intricate nature of PTMs, a more stringent site confidence score cutoff (>0.99) needs to be used for its comparable error rates with conventional methods. Nevertheless, these approaches bear the potential to find novel PTMs that are not included in any current database.
Comparatively, library-based approaches for PTM-DIA were often applied for proteome-wide profiling. Several studies have used the established library-based OpenSWATH and PEAKS workflows for N- and O-glycosylation analyses (10, 11). As an extension of OpenSWATH, Inference of PeptidoForms (IPF) (2) proposes an approach requiring more evidence (i.e., peptide query parameters) to assign a peptidoform confidence to each peak group, thereby increasing their identification confidence. Also, IPF has the potential to resolve identification conflicts when the m/z increment is below the width of the precursors’ isolation windows, which causes the “site-specific issue”. It has been used to resolve 12 oxidized peptidoforms caused by 4 PTMs within protein APOA1 (2).
Although the performances vary depending on the experimental settings and analysis strategies, PTM-DIA exhibits higher reproducibility (coefficient of variation <15%) and broader quantification capabilities (over four orders of magnitude) than PTM-DDA (12, 13). Currently, more than ten types of PTMs can be explored using PTM-DIA (2). Future studies may expand the software capabilities to allow the measurement and analysis of more PTMs.
Deep learning (DL) is becoming a valuable tool in analyzing proteoform data of highly complex structures and dimensionalities (14). Firstly, data-driven DL techniques are proven effective to predict spectral libraries (15). pDeep2 applies a transfer learning technique to predict phosphopeptides using the synthetic PTM data sets (16). DeepPhospho uses a deep neural network, trained with four large phosphoproteomics datasets, to generate in silico spectral libraries, which allowed the discovery of EGF-regulated signaling pathways and kinases (17). Secondly, the proteome-wide PTM sites identified using DIA-MS data could be used for DL-based prediction models (13). Future software developments could incorporate public DIA-MS data to boost prediction accuracy or support cross-validation analyses.
References
1. Smith LM, Kelleher NL, Consortium for Top Down P. Proteoform: a single term describing protein complexity. Nature methods. 2013;10(3):186-7.
2. Rosenberger G, Liu Y, Röst HL, Ludwig C, Buil A, Bensimon A, et al. Inference and quantification of peptidoforms in large sample cohorts by SWATH-MS. Nature biotechnology. 2017;35(8):781-8.
3. Thygesen C, Boll I, Finsen B, Modzel M, Larsen MR. Characterizing disease-associated changes in post-translational modifications by mass spectrometry. Expert Rev Proteomics. 2018;15(3):245-58.
4. Sidoli S, Lin S, Xiong L, Bhanu NV, Karch KR, Johansen E, et al. Sequential Window Acquisition of all Theoretical Mass Spectra (SWATH) Analysis for Characterization and Quantification of Histone Post-translational Modifications*[S]. Molecular & Cellular Proteomics. 2015;14(9):2420-8.
5. Krautkramer KA, Reiter L, Denu JM, Dowell JA. Quantification of SAHA-Dependent Changes in Histone Modifications Using Data-Independent Acquisition Mass Spectrometry. Journal of proteome research. 2015;14(8):3252-62.
6. Lawrence RT, Searle BC, Llovet A, Villén J. Plug-and-play analysis of the human phosphoproteome by targeted high-resolution mass spectrometry. Nature Methods. 2016;13(5):431-4.
7. Porter CJ, Bereman MS. Data-independent-acquisition mass spectrometry for identification of targeted-peptide site-specific modifications. Analytical and bioanalytical chemistry. 2015;407(22):6627-35.
8. Keller A, Bader SL, Kusebauch U, Shteynberg D, Hood L, Moritz RL. Opening a SWATH Window on Posttranslational Modifications: Automated Pursuit of Modified Peptides. Molecular & cellular proteomics : MCP. 2016;15(3):1151-63.
9. Bekker-Jensen DB, Bernhardt OM, Hogrebe A, Martinez-Val A, Verbeke L, Gandhi T, et al. Rapid and site-specific deep phosphoproteome profiling by data-independent acquisition without the need for spectral libraries. Nature Communications. 2020;11(1):787.
10. Sajic T, Liu Y, Arvaniti E, Surinova S, Williams EG, Schiess R, et al. Similarities and Differences of Blood N-Glycoproteins in Five Solid Carcinomas at Localized Clinical Stage Analyzed by SWATH-MS. Cell Reports. 2018;23(9):2819-31.e5.
11. Yang X, Wang Z, Guo L, Zhu Z, Zhang Y. Proteome-Wide Analysis of N-Glycosylation Stoichiometry Using SWATH Technology. Journal of Proteome Research. 2017;16(10):3830-40.
12. Sinitcyn P, Hamzeiy H, Salinas Soto F, Itzhak D, McCarthy F, Wichmann C, et al. MaxDIA enables library-based and library-free data-independent acquisition proteomics. Nat Biotechnol. 2021.
13. Wen B, Zeng W-F, Liao Y, Shi Z, Savage SR, Jiang W, et al. Deep Learning in Proteomics. Proteomics. 2020;20(21-22):e1900335-e.
14. Yue L, Zhang F, Sun R, Sun Y, Yuan C, Zhu Y, et al. Generating Proteomic Big Data for Precision Medicine. Proteomics. 2020;20(21-22):e1900358.
15. Ye Z, Vakhrushev SY. The Role of Data-Independent Acquisition for Glycoproteomics. Molecular & Cellular Proteomics. 2021;20:100042.
16. Zeng WF, Zhou XX, Zhou WJ, Chi H, Zhan J, He SM. MS/MS Spectrum Prediction for Modified Peptides Using pDeep2 Trained by Transfer Learning. Anal Chem. 2019;91(15):9724-31.
17. Lou R, Liu W, Li R, Li S, He X, Shui W. DeepPhospho accelerates DIA phosphoproteome profiling through in silico library generation. Nature Communications. 2021;12(1):6685.