This special issue launched by CNHUPO (HUPO in China) reviews the major advances in proteomics from 2001 (HUPO initiation) to 2021 (the 20th anniversary of HUPO). Jiaqi Zhang wrote the draft. Shanjun Chen, Junhong Xiao reviewed the language. I reviewed the scientific content. Permission to reproduce and modify the issue was granted. The original version (Chinese) could be referenced here. You could use the Language Switch icon at the top right of this page for assisted reading.
The original title: HUPO 蛋白质组学标准化项目 | 数据标准化挑战(1)

自1977年末端终止测序法(第一代DNA测序技术)诞生以来,学术界、工业界迫切希望谱写生命天书以深入解读人类生老病死的发展变化机制。2000年初,人类科学史上的伟大工程——人类基因组工作草图宣布绘制完成。随着这一重磅消息空袭全球,生命科学瞬间跃升为全民焦点话题,生物科学相关技术及产业更是如雨后春笋般涌现。
部分专家学者希望并承诺,研究人员将充分利用该项目产生的爆炸性数据,加速新药物疗法和诊断工具的开发进程。但事情并不像他们想得那样乐观。
首先,这些爆炸性数据并没有帮助彼时的制药公司把研究和开发药物的花费控制下来。因为尽管获得了大量数据,但在早期的研究中,研究人员无法通过基因片段破解基因表达调控机制;另一方面,大部分的基因变化是功能缺失引起的,我们很难通过研发药物来实现基因功能的重建。因此,科研人员转变思路研究基因的表达产物蛋白质,希望在蛋白质分子水平破解疾病发生发展机制。
所谓“生在基因,命在蛋白”,大部分人类疾病因蛋白质变化而引起,包括组成结构或功能的变化。蛋白质不仅可以构成生命——组建人类体内细胞结构;也可以毁灭生命——产生威胁生命的疾病。另一方面,与基因组相比,蛋白质具有动态性、时间性、空间性以及特异性等特点。这意味着,如果能对不同生理状态下的细胞、组织,以及正常和病理状态下的蛋白质组进行有效分析,确定疾病特异性蛋白质标记物,将能深化人类对疾病发生发展规律的认识,以及推动开发更加有效的治疗方法。
因此,全球研究机构投入大量科研资源开发高通量多样本制备方法、利用质谱等分析技术对蛋白质展开大量研究,并创造出海量数据。庞大的数据体量及复杂多维的信息,为数据分析、存储、计算带来了全新的挑战。不仅如此,数据记录标准及数据处理格式等因素的各异化更是将解密难度提升至另一令人望而生畏的维度。实现数据共享,使数据格式和分析方法标准化的切实需求。
一场蛋白质组学数据标准化之战,随之打响。
2002年,国际人类蛋白质组组织(Human Proteome Organization,HUPO)为此成立蛋白质组学标准化项目(Proteomics Standards Initiative, PSI,https://www.hupo.org/proteomics-standards-initiative/)。该项目旨在将蛋白质组学数据标准化,借此来实现数据比较、交换以及验证,并已得到数据核心提供者的广泛支持。此前虽然也有质谱仪供应商所开发及基于某些实验要求而特定开发的数据格式,但它们并不具有普适性。而HUPO-PSI开发的格式则是基于数据提供者、供应商、使用者等团队共同探讨开发的,更具普适性,在蛋白质组学领域的意义也不言而喻。

截止至今,HUPO-PSI先后创建了7个项目:分子相互作用(Molecular Interactions,MI)、质谱分析 (Mass Spectrometry,MS)、蛋白质翻译后修饰(Protein Modifications,MOD;似乎自2014年HUPO-PSI审查会议后不再活跃)、蛋白质分离(Protein Separation,PS;似乎自2014年HUPO-PSI审查会议后不再活跃)、蛋白质组学信息学(Proteomics Informatics,PI)固有无序蛋白质(Intrinsically Disordered Proteins,IDPs)、以及质量控制(Quality Control,QC),每个项目都有相应的工作组。我们将在下文逐一向大家介绍以上7个工作组的工作内容及进程。
1 分子相互作用 (Molecular Interactions, MI)
随着蛋白质组学的蓬勃发展,蛋白质分子相互作用已然成为生命科学领域的焦点课题。科研人员亦投入大量时间、精力对蛋白质分子相互作用网络性质深入研究,但因各实验室数据以不同形式(纸质或线上格式)记录于数据库、作者网站或出版物等资源中,导致跨实验数据整合、全球数据分享等工作十分受阻。
HUPO-PSI就以上问题成立蛋白质分子相互作用工作组,其核心工作包括:(1)从期刊文章、作者网站及公共数据库等资源中收集、整理蛋白质分子相互作用数据,并进行注释和可视化呈现;(2)确定并应用标准数据格式储存数据使其统一化,以提高用户群体对分子相互作用数据的可及性。这些工作不仅可以实现数据的系统性采集、提高重复性使用和促进数据交换,也催生了新的分析工具。例如,通过整合蛋白质组学与基因组学的数据信息能揭示更多新生物学知识,以促进分子相互作用网络式分析以及与多组学的交互分析,这是单一组学数据分析难以企及的。
2002-03年期间,研究人员开始开发基于可扩展标记语言(Extensible Markup Language,XML)的文件格式以存储质谱数据。该格式基于文本格式,不仅适用于任意操作系统,亦方便编程语言读取。2004年,HUPO-PSI开发标准交换格式PSI-MI XM 1.0版,并得到软件工具开发工程师和数据提供者的广泛支持。该格式利用一系列软件工具对数据进行整合分析、可视化处理,并最终实现数据共享。HUPO-PSI曾明确表示,PSI-MI格式将渐进发展。因此,PSI-MI XM 1.0版本只关注蛋白质间的相互作用,对定量参数的支持非常有限。
2005年12月5日,为满足用户、数据库组和数据提供者的更高要求,HUPO-PSI将PSI-MI XM升级至2.5版本。该版本的主要特点是扩大了相互作用物的类型范围,细化了对实验背景环境及相互作用蛋白的特征描述,并增加了动力学和数据模型间的相互作用参数。

▲ Samuel Kerrien带领团队发表文章Broadening the horizon – level 2.5 of the HUPO-PSI format for molecular interactions
逐渐地,PSI-XML 2.5因难以满足不断发展的研究需求走向其应用“天花板”:无法解析通过整合多个实验而得到的蛋白质间相互作用推断,如变构/合作相互作用 (Allosteric/cooperative interactions) 、蛋白质复合体 (Protein complexes)和动态相互作用(Dynamic interactions)。
2016年,数据生产者、使用者、提供者及工具开发者随后合作开发出PSI-MI XML 3.0(该版本沿用至今)。他们建议研究人员继续使用PSI-MI XML 2.5作为实验数据的主要交换格式(在PSI-X ML 3.0开发前,PSI-X ML 2.5已更新至2.5.4版,并沿用至今),而PSI-MI XML 3.0则用于处理更复杂的数据类型,以及制表符分隔的文本格式MITAB 2.5、MITAB 2.6和MITAB 2.7中。
2 质谱分析 (Mass Spectrometry, MS)
质谱拥有百年发展历史,科学家们为满足不同研究需求而开发出各式各样的质谱分析法及仪器设备,随之而来的数据处理软件及数据格式等纷繁多样,使得数据整合陷入窘境。同时,质谱数据在许多层面上存在差异,造成差异的因素亦非常多样,包括数据转换造成的差异——从原始数据(Raw data)到峰值列表(Peak lists),从肽段识别到蛋白识别等过程。
因此,质谱工作组需要解决两个主要问题:(1)将大部分商业硬件的数据输出格式标准化,使数据可以被任意搜索引擎读取或存储于兼容的数据库中;(2)开发质谱图标准输出格式,以及肽段、蛋白质鉴定和修饰的通用术语表(Controlled vocabulary,CV),这将避免因使用不同搜索引擎而带来的差异化结果。
2003年,HUPO-PSI质谱数据标准化工作组在当年的HUPO大会上公布mzData交换格式的初版本,以便研究人员进行数据交换。2004年,系统生物学研究所蛋白质组中心(Proteome Center at the Institute for Systems Biology)开发mzXML格式,以简化数据处理。mzData是基于限制性词汇表设计的,可随着质谱技术的发展而更新。而mzXML具有严格的设计架构,可描述辅助信息,根据质谱技术发展情况进行相应修订。在这两种格式都被广泛应用的同时,研究人员意识到,本质相同的东西(都是基于XML格式开发的)如果存在两种不同格式,将导致不必要的混乱和额外的编程工作。
2006年,HUPO-PSI根据mzXML和mzData格式的设计原则,以及仪器和软件供应商、数据存储库、终端用户的建议,对以上两种格式进行整合并确定标准格式的基础需求:(1)简化标准格式;(2)确保相同信息仅有一个编码方式;(3)支持mzXML和mzData的所有功能;以及(4)格式发布前进行严格的审核验证。
2008年6月,满足以上要求的mzML 1.0.0标准格式顺利诞生。随着该格式的广泛应用,其缺点也逐渐暴露——无法对母离子扫描(Precursor Ion Scan)和中性损耗扫描(Neutral loss scans)提供充分支持,以及SRM(Selected Reaction Monitoring)文件的膨胀问题。
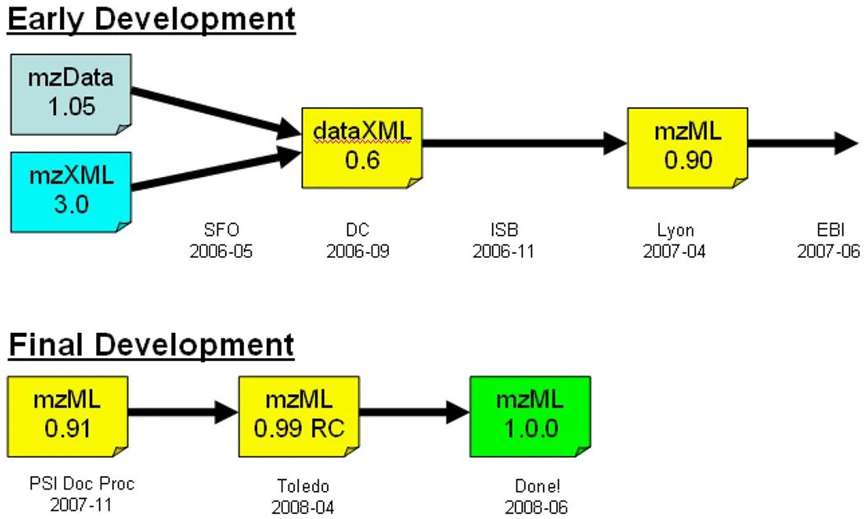
▲mzML 1.0.0发展历史6

▲mzML文档包括质谱图以及相关元数据信息,如样品名称、处理软件名称、设备类型、数据处理信息以及数据采集信息等6
PSI-MS工作组与发现以上问题的研究人员合作纠正了这些缺陷以及其他小问题,并于2009年6月发布了mzML 1.1.0版本(该版本沿用至今)。该版本也被广泛证明为是一种可靠格式——在可完美适应质谱技术渐进式发展的同时,也为适应新技术的数据编程扩展工作打下良好基础。
另外,随着SRM靶向质谱技术的出现及广泛应用,HUPO-PSI MS工作组设计了专属的实验数据交换、传输标准格式——TraML 1.0.0,并公布于2011年(该版本沿用至今)。该格式的设计原理与mzML相似,都是通过限制性词汇表进行编码元数据,可被许多工具解析。与mzML不一样的是,TraML并不是为储存原始质谱数据而设计,而是用于确定质谱仪选择碎裂的离子、编码过渡列表以及与这些列表相关的元数据。

▲TraML是公共或私人数据库中过渡列表、SRM设计、分析软件以及仪器控制软件之间的通用标准交换格式8

▲TraML格式示意概述图8
3 蛋白质组学信息学 (Proteomics Informatics, PI)
随着质谱分析技术的发展,蛋白质组学分析仪器精度不断提高,由此获得的数据也更多。但研究人员无法仅依靠人工对如此庞大的质谱信息进行深度分析,并揭示蛋白质分子特征,这些数据需要采用专业的信息学方法进行分析处理。但由于研究人员读取、导出数据的软件不同,蛋白质和多肽的鉴定结果以及辅助信息(如搜索参数)亦以不同格式呈现。
为攻克这一难题,HUPO-PSI成立蛋白质组学信息学工作组,以制定标准化数据格式及相关术语表。这将促进下游数据库共享分析、存档,包括通过软件对蛋白质、多肽进行鉴定量化,以及蛋白质组学与其他组学数据(如蛋白质基因组学分析)的系统性分析。
2009年,HUPO-PSI 开发了mzIdentML 1.0格式(已不适用)以标准化蛋白质与肽段的鉴定分析结果,以及规范化方法、参数和质量指标元数据的储存格式。2011年,HUPO-PSI修正了mzIdentML 1.0格式的不足,使其能够被稳定应用,并进阶为mzIdentML 1.1版本。该格式自发布以来受到数据软件的广泛支持,许多分析工具将它纳入其中,如MS-GF+,Mascot(Matrix Science,London,UK,from version 2.4),及ProteinPilot (SCIEX,Framingham,MA,from version 5.0) 等。
2013年,研究人员为定量结果建立了一个单独的格式——mzQuantML,并在第二年开发了一种更简易的鉴定和定量结果采集格式——mzTab 1.0(该版本沿用至今)。研究人员认为,该格式能够极大地促进全球蛋白质组学数据共享,并成为定量蛋白质组学信息学发展的基础。因为该格式不仅可以将数据文本格式存储于表格中,使其被直接加载到Excel表格或统计软件中;还可以无缝提交至PRIDE、MassIVE、jPOST、iProx等数据库中。
此外,随着蛋白质组信息学的高速发展,mzIdentML 1.2格式于2017年被成功开发以满足更广泛的研究需求(该版本沿用至今):(1)确定翻译后修饰或化学修饰位置的相关分数;(2)提高肽段匹配度;(3)识别交联肽(Cross-linked peptides);以及(4)支持蛋白质基因组学分析。该版本也对多肽从头测序(De novo sequencing of peptides)、图谱数据库(Spectral library searches)和蛋白质推断(Protein inference)提供了更大的支持。同时,为简化应用者从1.1到1.2版本的过渡及确保其应用稳定性,研究人员仅对mzIdentML的底层XML模式进行细微修改;格式规范、实施指南、术语表(包含逾2700个术语和定义)和验证软件也都有了一些明显的更新。

▲ Andrew R.Jones团队发表文章The mzIdentML Data Standard Version 1.2, Supporting Advances in Proteome Informatics
参考资料
1.May, M. (2018). Translating big data: The proteomics challenge. Science, 360(6394), 1252-1254.
2.Kerrien, S., Orchard, S., Montecchi-Palazzi, L., Aranda, B., Quinn, A. F., Vinod, N., … & Hermjakob, H. (2007). Broadening the horizon–level 2.5 of the HUPO-PSI format for molecular interactions. BMC biology, 5(1), 1-11.
3.Alonso-López, D., Ammari, M., Bradley, G., Campbell, N. H., Ceol, A., Cesareni, G., … & Orchard, S. (2018). Encompassing new use cases-level 3.0 of the HUPO-PSI format for molecular interactions. BMC bioinformatics, 19(1), 1-8.
4.Deutsch, E. W., Orchard, S., Binz, P. A., Bittremieux, W., Eisenacher, M., Hermjakob, H., … & Jones, A. R. (2017). Proteomics standards initiative: fifteen years of progress and future work. Journal of proteome research, 16(12), 4288-4298.
5. LeDuc, R. D., Deutsch, E. W., Binz, P. A., Fellers, R. T., Cesnik, A. J., Klein, J. A., … & Vizcaíno, J. A. (2022). Proteomics Standards Initiative’s ProForma 2.0: Unifying the Encoding of Proteoforms and Peptidoforms. Journal of proteome research, 21(4), 1189-1195.
6.Deutsch, E. W. (2010). Mass spectrometer output file format mzML. In Proteome bioinformatics (pp. 319-331). Humana Press.
7.Martens, L., Chambers, M., Sturm, M., Kessner, D., Levander, F., Shofstahl, J., … & Deutsch, E. W. (2011). mzML—a community standard for mass spectrometry data. Molecular & Cellular Proteomics, 10(1).
8. Deutsch, E. W., Chambers, M., Neumann, S., Levander, F., Binz, P. A., Shofstahl, J., … & Brusniak, M. Y. (2012). TraML—a standard format for exchange of selected reaction monitoring transition lists. Molecular & Cellular Proteomics, 11(4).
9. Vizcaíno, J. A., Mayer, G., Perkins, S., Barsnes, H., Vaudel, M., Perez-Riverol, Y., … & Jones, A. R. (2017). The mzIdentML data standard version 1.2, supporting advances in proteome informatics. Molecular & cellular proteomics, 16(7), 1275-1285.