English title: SEQUEST: Pioneer of the Year 1994
The original version of this article was published in the HUPO Official Wechat. Dr. Luyao Liu from Dalian Institute of Chemical Physics, CAS helped with the revision. I made several edits in this version.
原文标题:An Approach to Correlate Tandem Mass Spectral Data of Peptides with Amino Acid Sequences in a Protein Database;
原文链接:https://pubs.acs.org/doi/10.1016/1044-0305(94)80016-2


一作:Jimmy Eng;通讯:John Yates.
SEQUEST是由Jimmy Eng和John Yates领衔、在1994年完成设计的、解析串联质谱数据、计算得出生物氨基酸序列的技术工作。25年过去了,如今这项工作的原始论文被引用7108次(截至2021年5月14日),是美国质谱协会杂志(Journal of the American Society for Mass Spectrometry, JASMS)创刊以来引用最多的科学论文。就影响力而言,作为自下而上蛋白质组学检索程序的开山鼻祖,SEQUEST不仅激励了更多的资源投入和大胆尝试,当今如火如荼的各种蛋白质组软件和算法,或多或少地都受之启发;还推动了基于质谱的蛋白质组领域的发展,使之成为本世纪最激动人心的生物技术之一。
SEQUEST is a technical effort led by Jimmy Eng and John Yates, designed in 1994, to resolve tandem mass spectrometry data and compute biological amino acid sequences. Twenty-five years since its publication, the original study was cited 7,108 times (by May 14, 2021), making it the most cited scientific paper in the Journal of the American Society for Mass Spectrometry (JASMS) since inception. SEQUEST, as the pioneer of bottom-up proteomic search programs, has not only inspired more resources and bold experimentation but also inspired the various proteomic software and algorithms that are in full swing today. It has also driven the development in the field of mass spectrometry (MS)-based proteomics, one of the most exciting biotechnologies of the century.
笔者将尝试对这篇生物质谱数据分析的“老大哥”进行解读,重点放在方法的搭建和验证;再发表一些拙见,以期抛砖引玉。
The editors will try to interpret this “big brother” of biological mass spectrometry data analysis, focusing on the construction and validation of the method. Then, they will show some humble opinions, with a view of throw out a brick to attract a jade.
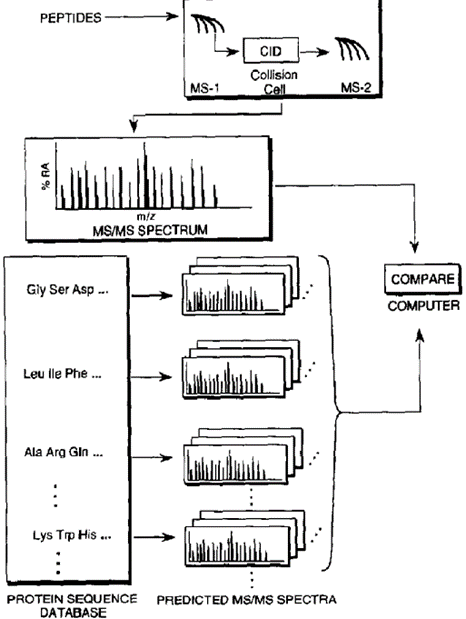
概况的说,SEQUEST分析方法分成四步(图一)。第一步,对质谱数据进行压缩;第二步,通过比对蛋白质数据库(database)与实验质谱数据在分子质量层面的信息,匹配(compare)可能的多肽序列;第三步,将从数据库中得到的序列的预测片段离子与质谱信息进行比较,从而产生最佳匹配序列表;这个序列被用于进行打分和统计学运算,进入最后一步,也就是得到分析结果。
Generally, the SEQUEST analysis method is divided into four steps (Figure 1). The first step begins with computer reduction of the mass spectrometry data; The second step is to match the possible peptide sequences by comparing the molecular weight information of the protein database with the experimental mass spectrometry data; The third step is to compare the predicted fragment ions of the sequences obtained from the database with the mass spectrum information to generate the best matching sequence table. This sequence is used to perform scoring and statistical operations to get to the final step, which will show the analysis results.
第一步:串联质谱数据的压缩(Tandem mass spectrometry data reduction)
Step 1:Tandem mass spectrometry data reduction
SEQUEST通过以下三步对得到的谱图进行简化:
SEQUEST simplifies the obtained spectra by the following three steps:
1) 首先,将碎片离子的质荷比转换成标称值(四舍五入为整数),以期提高检索效率;
First of all, convert the mass-to-charge (m/z) ratio of fragment ions to nominal values (rounded to integers) to improve database searching efficiency.
2) 接下来,移除二级谱图中前体离子质荷比范围前后5个单位内的信号,以避免未打碎的前体离子造成的信号干扰或错配。
Next, Remove the signal within 10 units around the precursor ions in the MS2 to avoid signal interference or mismatch caused by unfragmented precursor ions.
3) 最后,只保留并归一化谱图中丰度前x位的碎片离子峰,而删去其余碎片离子峰,以消除谱图中的噪音离子并减少要考虑的离子数量。x ∈{100,200,500}
At last, only the top x most abundance fragment ions in the spectrum are retained and normalized, while the remaining fragment ions are deleted to eliminate noise in the spectrum and reduce the number of ions to be considered. x ∈{100,200,500}
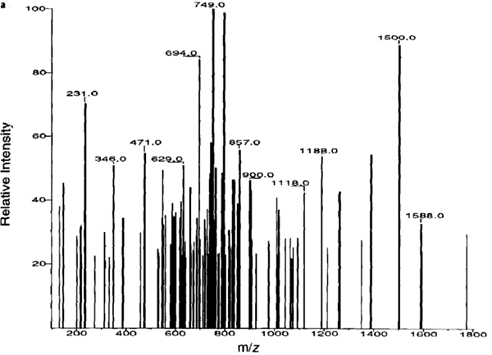
多肽DLRSWTAADTAAQISQ串联质谱数据经过压缩后的谱图如图二。
The compressed spectrum of the peptide DLRSWTAADTAAQISQ tandem mass spectrometry data is shown in Figure 2.
第二步:多肽序列的搜索(Search method)
Step 2: Search method
这一步是要注释谱图,也就是匹配谱图中的碎片离子和数据库中的多肽序列。SEQUEST开创性地采用了数据库搜索的方法:利用由人类基因组计划的核苷酸链数据来推导得到的氨基酸链组成的数据库,将其中氨基酸的分子质量从N端到C端一一加和,直到其能与待分析的谱图中给出的质荷比相匹配。在这里,匹配的误差被设定为在正负0.05%、或者一个单位以内。数据库中碎片离子的质荷比通过如下两个简单公式来计算:
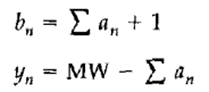
其中,an是氨基酸的质量,bn是b型离子,yn是y型离子。此外,SEQUEST还考虑了氨基酸的化学修饰(chemical modifications),通过不同的修饰对应于不同的氨基酸质量变化来计算多肽的质量,并被先验地存储在搜索引擎内。然而,作者为了原始算法的简易性和普适性而不考虑糖基化和磷酸化修饰,原因分别是糖基化在碰撞诱导解离(collision-induced dissociation, CID)中会影响多肽的碎裂规律和磷酸化表现了极大的异质性。
This step is to annotate the spectra, that is, to match the fragment ions in the spectra with the peptide sequences in the database. SEQUEST uses the database search method as a pioneer: Using a database of amino acid sequences derived from nucleotide chain data of the Human Genome Project, the molecular masses of the amino acid sequences are summed one by one from the N to the C-terminus until they can match the m/z given in the spectrum. The tolerance is set at ± 0.05% or within one unit. The m/z of the fragment ions in the database is calculated by two simple equations as follows:
an is the mass of the amino acid, bn is the b ion, and yn is the y ion. In addition, SEQUEST takes chemical modifications of amino acids into consideration. The mass of the peptide is calculated by different modifications corresponding to different amino acid mass changes and is stored a priori in the search engine. However, the authors disregarded glycosylation and phosphorylation for simplicity and generalizability of the original algorithm, because glycosylation affects the fragmentation pattern of the peptides in collision-induced dissociation (CID) and phosphorylation shows great heterogeneity, respectively.
第三步:多肽序列的打分(Scoring method)
Step 3: Scoring method
SEQUEST通过打分(scoring)的机制来实现多肽序列的鉴定。
SEQUEST enables the identification of peptide sequences through a scoring mechanism.
首先,对于第二步中得到的匹配结果,也就是一张张二级谱图对应的可能性碎片离子,记其离子数目为ni和丰度im;
First of all, for the matching results obtained in the second step, that is, the possible fragment ions in MS2, mark the number of ions as ni and their abundance im ;
并且,设置打分β以表征这些碎片离子的连续性(The continuity of an ion series),数值0.075;
Set a scoring β to characterize the continuity of the fragment ions with a value of 0.075;
如果出现了His,Tyr,Trp,Met和Phe的亚胺离子,再设置打分ρ,数值0.15;
If imine ions of His, Tyr, Trp, Met, and Phe are present,set the scoring ρ with a value of 0.15;
最后,数据库中该多肽的可能碎片离子总数计为nt。
At last, count the total number of possible fragment ions of the peptide in the database is as nt.
将如上的参数和打分合并在如下公式中、以求得总打分Sρ:
Combine the above parameters and scores in the following formula to calculate the total score Sρ :
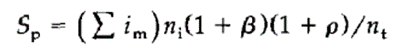
为了验证这个打分,作者采集了57组质谱数据做分析,得到如下结论:
To verify this scoring, the authors analyzed 57 mass spectrometry data and reached the following conclusions:
1) 通过相同的多肽在不同组实验中得到的数据的分析,确定了这个打分程序的高重复性;
The high reproducibility of this scoring method was determined from different experimental groups with the same peptide;
2) 该打分程序没有考虑因不同电荷态引起的多个碎片离子,因此对其中一种电荷态的识别结果往往会变差;
The scoring method does not take multiple fragment ions into consideration due to the different charge states, and therefore the identification of one of the charge states tends to be poor;
3) 该打分程序对于短肽(不长于四个氨基酸)的鉴定,准确率不高;短肽的识别结果往往排不到可能结果中的前五百位;
The scoring method is not very accurate for identifying short peptides (no longer than four amino acids); identification of short peptides often does not rank in the top 500 of possible results.
4) 该打分程序难以评估结果的假阳性(false positives)。
The scoring method makes it difficult to assess the false positives of the results.
藉此,作者们设计了第二项独立的打分程序以优化结果。
In such a case, the authors designed a second independent scoring procedure to optimize the results.
第四步:多肽序列的互相关分析(Cross-correlation analysis)
Step 4: Cross-correlation analysis
SEQUEST借鉴了先前被用于红外谱图库中谱图相似性比较的互相关分析,即通过将一个信号转换成另一个信号来测量两个信号的相干性,在这里用于进行数据库谱图数据和实验谱图数据的比较。
SEQUEST draws on the cross-correlation analysis previously used for spectral similarity comparisons in infrared library search, where the coherence of two signals is measured by converting one signal to the other, and is used here to perform comparisons between database spectral data and experimental spectral data.
1) 首先处理由数据库推测得到的二级谱图。由于对于通过数据库推断得到的碎片离子,其强度(intensity)几乎无法预测,于是SEQUEST进行了如下假设:将所有b、y离子强度均调整为50%;而将与b、y离子质荷比相差在正负一个单位以内的离子强度调整为25%;而对于在三重四极谱图中很常见的水、氨和一氧化碳(a型离子)的中性损失,SEQUEST赋其值为10%。这样,推测得到的谱图既包括了碎片离子强度的信息,又将碎片离子强度对相关性的影响大大降低,只作为辅助条件(图三);
The MS2 spectra obtained from the database prediction are processed first. Since the intensity of the fragment ions obtained by database inference is almost impossible to predict, SEQUEST made the following assumptions: adjust the intensity of all b and y ion to 50%; and set the intensity of ions whose m/z differs from b and y ions within ± one unit to 25%; for the neutral losses of water, ammonia and carbon monoxide (a-type ions), which are common in triple quadrupole spectra, SEQUEST assigned a value of 10%. In this way, the inferred spectrum not only includes the information of the ionic strength of fragments but also greatly reduces the effect of the ionic intensity of fragments on the correlation, which is only an auxiliary condition (Figure 3)
2)对于压缩后的实验二级谱图,SEQUEST先将采集质量范围分成10个均匀的窗口,然后将每个窗口内的离子归一化(normalize)到50%(图四):
For the compressed experimental MS2 spectra, SEQUEST first divided the mass acquisition range into 10 uniform windows and then normalized the ions within each window to 50% (Figure 4):
3)对两张预处理后的二级谱图做互相关函数分析:
Cross-correlation analysis of the two pre-processed secondary spectra:
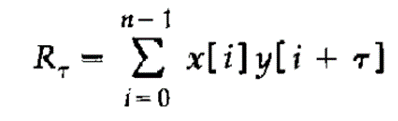
xi和yi分别代表重构的数据库谱图和实验谱图信号。它们经转换成为一组离散傅里叶变换对(Fourier transform pairs)先后进行了包括快速傅里叶变换、复共轭乘积,以及乘积逆变换等运算;τ是两个谱图信号之间的位移值。τ = 0时表示没有偏移,互相关函数达到最大值;Rτ的分布如下图(图五)所示。
xi and yi represent the reconstructed database spectra and experimental spectra signals, respectively. They are transformed into a set of discrete Fourier transform pairs that successively perform operations, including the fast Fourier transform, the complex conjugate product, and the product-inverse transform; τ is the value of the displacement between the two spectrogram signals. τ = 0 indicates no offset and that the inter-correlation function reaches its maximum value; The distribution of Rτ is shown in the following figure (Figure V).
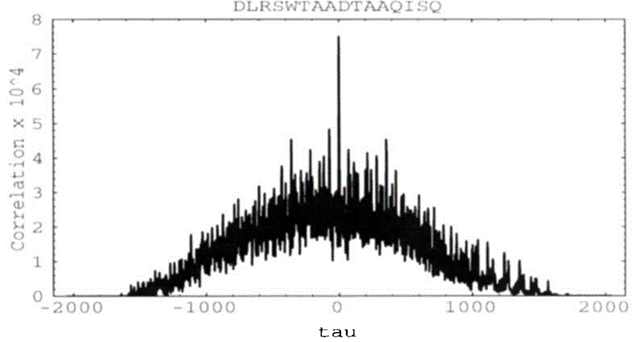
该步骤生成的总打分Cn由τ = 0时的函数值减去-75 < τ< 75范围内函数值的平均值,并归一化到1.0后得到。由于对于每一张实验谱图,我们都因为多张数据库谱图的匹配而生成了多个结果,因此多肽对于此打分程序的最佳结果(Cn最高)和次佳结果(Cn次高)之间的差异可以体现错误发现率。作者通过对已知序列的多肽进行评估发现,当最佳结果和次佳结果的Cn的差异大于0.1时,通常最佳结果是正确的(图六);Cn评分对于许多小肽的鉴定有益。
The total scoring Cn generated in this step is obtained by subtracting the average of the function values in the range -75 < τ < 75 from the function values at τ = 0, and normalizing to 1.0. Since for each experimental spectrum we generated multiple results due to the matching of multiple database spectra, the difference between the best result (highest Cn) and the next best result (second highest Cn) for this scoring procedure for peptides can reflect the false discovery rate. The authors found by evaluating peptides with known sequences that the best result was usually correct when the difference between the Cn of the best and the second-best result is greater than 0.1 (Figure 6). The Cn score is useful for the identification of many small peptides.
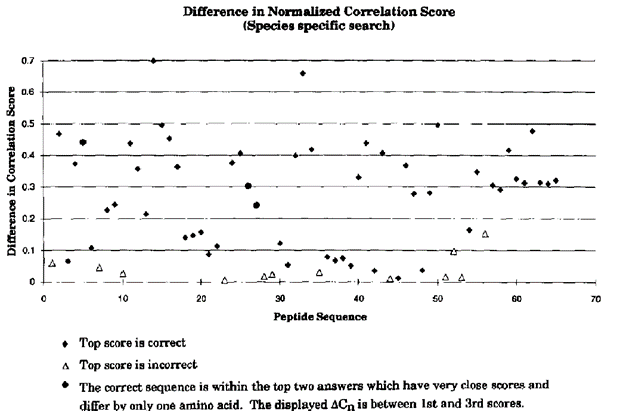
作者评估了改变匹配容忍度带来的影响,发现即使增大到3个单位,也就是囊括了更多可能性氨基酸的匹配,也不会对结果的正确性带来大的变化;神来之笔则是对不同数据库的使用做了比较,并证明了从整体来看,用物种特异性数据库比用全基因数据库(Genepept)的检索更准确。
The authors evaluated the impact of changing the match tolerance and found that it did not make a significant difference to the correctness of the results even if it was increased to three units, i.e., including more possible amino acid matches. The magic formula compared the use of different databases and demonstrated that, overall, searches with species-specific databases are more accurate than those with gene-wide databases.
最后,作者将SEQUEST分析方法应用在三种代表性的生物学问题中:
Finally, the authors applied the SEQUEST to three representative biological problems.
1)Ⅱ类主要组织相容性分子(major histocompatibility molecules, MHC)多肽的鉴定。Identification of class II major histocompatibility molecules (MHC) peptides.
Ⅱ 类MHC蛋白所产生的肽大多来自于外源性/内源性血清蛋白,或发生配位的细胞处理腔室中已知的蛋白,也就是说它们的基因序列大都已知。作者先通过液相色谱分出HLA DRB*0401 Ⅱ类MHC分子的50个馏分,其中30个馏分在220 nm处有紫外吸收,表明了肽的存在。作者对这30个馏分做LC-MS分析,并从中挑选出信噪比大于3/1且带有双电荷的信号肽离子作为分析对象,通过将容忍度设为1个单位,检索全基因数据库后,共鉴定出384,398个不同的氨基酸序列。
Most of the peptides produced by class II MHC proteins are derived from exogenous/endogenous serum proteins or from proteins known in the cellular processing compartment where the alignment occurs, meaning that most of their gene sequences are known. The authors first fractionated 50 fractions of HLA DRB*0401 class II MHC molecules by liquid chromatography (LC), 30 of which had UV absorption at 220 nm, indicating the presence of peptides. The authors did LC-MS analysis of these 30 fractions and selected signal-to-noise ratios greater than 3/1 and doubly charged signal peptide ions for analysis. 384,398 different amino acid sequences were identified by searching the Genpept database and setting the tolerance to 1 unit.
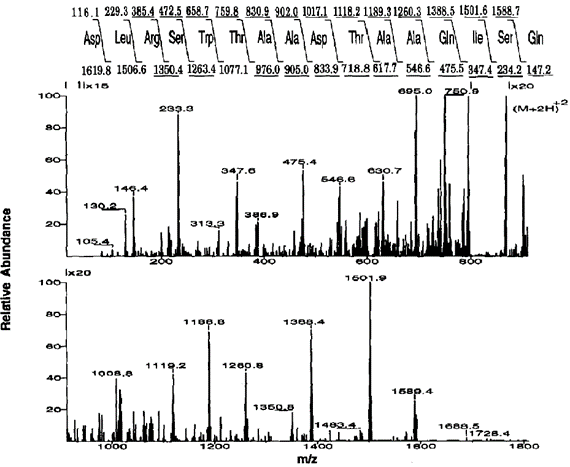
以质荷比为868的一个多肽的碎片离子谱图为例(图七),经过打分程序后,DLRSWTAADTAAQISQ和DLRSWTAADTAAQISK被认为是最概然的结果:都产生了相同的互相关参数和分数。由于Q(谷氨酰胺,Gln)和K(赖氨酸,Lys)作为序列的末位,不太可能在低能轰击下从肽链中断裂;因此作者推断正确的序列应该是DLRSWTAADTAAQISQ,其质量增加的42个单位被认为是带有一个游离氨基的乙酰化修饰导致。第一个应用表明看SEQEUST对实验数据的预测本领大大简化了抗原鉴定。
Taking the fragment ion spectrum of a peptide with an m/z of 868 as an example (Figure 7), DLRSWTAADTAAQISQ and DLRSWTAADTAAQISK were considered to be the most probable results after the scoring procedure: both processed the same correlation parameters and scores. Q (glutamine, Gln) and K (lysine, Lys) are unlikely to be broken from the peptide chain by low-energy bombardment due to their position as the end of the sequence: the authors therefore deduced that the correct sequence should be DLRSWTAADTAAQISQ, who’s increased 42 units in mass are thought to result from acetylation with a free amino group. The first application shows that the predictive power of SEQEUST on experimental data greatly simplifies the identification by the antigen-antibody reaction.
2) 细胞样本的多肽序列鉴定。Peptide sequence identification of cell samples.
作者对于从细胞中获得的蛋白/多肽混合物(protein complex)而不是单个多肽进行分析。由于不再需要先验的知识,这将直接有助于对于基因组/cDNA序列翻译的蛋白序列进行系统学的鉴定。作者用胰蛋白酶分别从大肠杆菌和大脑链球菌细胞中提取出蛋白多肽,做完一级质谱后挑选最高丰度的多肽离子(一般至少有10个氨基酸长度),采集其二级质谱数据。SEQUEST检索得到的序列与实际结果(手动检查得到的序列)吻合较好。并且,由于是对混合物的质谱鉴定,其丰度信息被部分保留,因此SEQUEST可以通过氨基酸序列的丰度来推测对应的蛋白数目。
The authors analyzed the protein complexes obtained from cells rather than a single peptide. Since a priori knowledge is no longer required, this will directly contribute to the phylogenetic identification of protein sequences translated from genomic/cDNA sequences. The authors extracted protein peptides from E. coli and S. cerevisiae cells using trypsin, respectively They selected the most abundant peptide ions (generally at least 10 amino acids in length) in MS1 to collect their MS2 data. The sequences obtained from the SEQUEST search matched better with the actual results (sequences obtained by manual inspection). And, since it was a mass spectrometric identification of the mixture, its abundance information is partially preserved, SEQUEST could infer the number of proteins from the amino acid sequence abundance.
3) 单一蛋白样本的多肽序列鉴定。Peptide sequence identification of single protein samples.
作者还对糖基化天冬酰胺酶的羧甲基化重链(S-carboxymethylated heavy chain of glycoasparaginase)水解后的多肽进行了分析。SEQUEST打分中最优结果和次优结果的差异很大,表明SEQUEST在这项应用中的匹配度较高。值得一提的是,SEQUEST每次搜索单个蛋白仅需要5秒;胰蛋白酶的水解并不会影响序列鉴定,即可以很好地实现自下而上推断蛋白;最后,理论上少量的污染蛋白、异构体、或不完全的水解也都影响不了SEQUEST分析流程。
The authors also analyzed the peptides after hydrolysis of the S-carboxymethylated heavy chain of glycoasparaginase. The considerable difference between the best and second-best results in SEQUEST scoring indicates that SEQUEST is a good match for this application. It is worth mentioning that SEQUEST takes only 5 seconds to search for a single protein each time; trypsin digestion does not affect sequence identification, that is to say, the bottom-up inference of proteins can be well achieved; finally, small amounts of contaminating proteins, isomers, or incomplete hydrolysis would theoretically not affect the SEQUEST analytical process.
讲到这里,一些读者们是不是已经云里雾里了?别急,课代表出来总结SEQUEST分析的知识点了:
Till now, some of readers may got confused. Don’t worry, here are the key points:
首先压缩数据,避免一级二级离子之间的干扰和噪音;
First of all, compress the data to avoid the interference and noise between MS1 and MS2 ions;
接着注释数据,把实验二级谱图和可能的序列给匹配出来;
Secondly, annotate the MS2 and match it with the possible sequences;
随后对匹配结果进行第一项打分Sρ,用于评估匹配度;
Thirdly, score Sρ for matching results to assess the matching degree;
最后对匹配结果进行第二项打分Cn,用于评估相关性。
At last, score Cn for matching results to apply the matching results for cross-correlation analysis.
作者通过对单一多肽、单一细胞全蛋白和单一蛋白序列的鉴定,给出了SEQUEST方法的应用方向。
The authors gave directions for the application of the SEQUEST through the identification of single peptides, total proteins from single-cell organisms, and single protein sequences.
这套方法既结合了当年最尖端的技术,如求互相关性的快速傅里叶变换(fast Fourier transform, FFT);又融入了作者在对质谱数据深入理解后的大胆假设,如对数据进行的系统归一化处理和多项经验打分权重等。值得提及的是,1994年也是数据依赖型采集(data-dependent acquisition)的诞生元年,SEQUEST也是最早使用DDA数据的工作之一。
This approach combines both the most sophisticated techniques of the day, such as the fast Fourier transform (FFT) for mutual correlation, with the authors’ bold assumptions based on a deep understanding of the mass spectrometry data, such as systematic normalization of the data and multiple empirical scoring weights. It is worth mentioning that 1994 was also the birth year of data-dependent acquisition (DDA), and SEQUEST was one of the first efforts to use DDA data.