来自德国弗莱堡大学的研究者们日前于BioRxiv上公开了他们基于Bruker的MALDI质谱timsTOF fleX进行PRM靶向分析多肽的研究工作,通过开发并优化一种基于捕获离子迁移谱(TIMS)的PASEF技术进行MALDI MS/MS成像的新方法iprm-PASEF,实现了对多条酶解肽段进行同时的空间分析,从而为空间蛋白质组学(Spatial Proteomics)提供了潜在的新方案。论文链接:https://www.biorxiv.org/content/10.1101/2024.11.08.622662v1.full.pdf;标题:Spatial Proteomics by Trapped Ion Mobility supported MALDI MS/MS Imaging: A First Glance into Multiplexed and Spatial Peptide Identification。

MALDI成像能够提供分子在组织中的空间分布信息,但在肽段的原位鉴定方面仍存在挑战,这主要是因为传统的MALDI成像限于MS1水平,缺乏MS2碎片谱来确定肽段序列;如今虽然MALDI也能兼容二级质谱,但一方面由于缺乏LC的分离而不可避免地导致采集的二级谱图中存在大量等重肽;另一方面大量的MALDI系统都只支持“一颗子弹消灭一个敌人”:一个激光束打一个母离子,因此无法做到检测数目上的高通量。
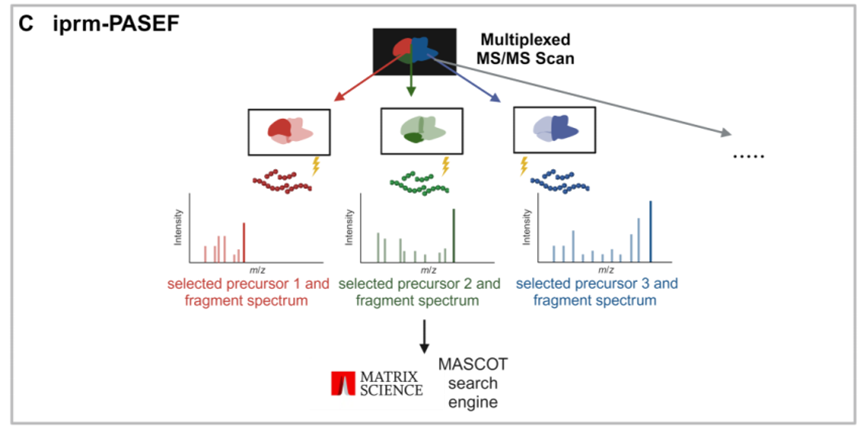
iprm-PASEF是Bruker公司为其timsTOF MALDI系列仪器开发的新型采集模式,通过软件调谐实现了在同一激光束下根据TIMS的分离对多个母离子进行分析。该采集模式已经应用于如脂质和代谢物的多重分析中(笔者注:感兴趣的读者也可以参考Bruker官网视频内容:https://www.bruker.com/zh/news-and-events/webinars/2024/confidence-in-spatial-identifications-using-iprm-pasef-on-the-timstof-flex.html)。
在本项工作中,研究者们首次尝试将该方法应用到多肽分析中,首先在MALDI TIMS MS1模式下对样品进行扫描,获得空间信息并生成一个前体列表;然后在iprm-PASEF模式下,选取最多五条多肽进行成像,获取MS2碎片谱;最后即是对谱图的注释。研究例中,使用50μm的MS1分辨率进行空间分析,并用100μm的分辨率获得MS2谱,平均耗时不到100分钟即可完成1 cm²区域、多达2万多个像素点的扫描。研究结果显示,MALDI相较于做对比的ESI源对于获取多肽的m/z值误差小于5 ppm,1/K0值误差小于0.01 V•s/cm²,表明LC-ESI和MALDI成像的肽段迁移特性具有高度一致性。
iprm-PASEF已经来了,idia-PASEF还会远吗?让我们把窗户打开(前体离子窗口扩大)并翘首以盼。