This special issue launched by CNHUPO (HUPO in China) reviews the major advances in proteomics from 2001 (HUPO initiation) to 2021 (the 20th anniversary of HUPO). Jiaqi Zhang wrote the draft. Shanjun Chen, Junhong Xiao reviewed the language. I reviewed the scientific content. Permission to reproduce and modify the issue was granted. The original version (Chinese) could be referenced here. You could use the Language Switch icon at the top right of this page for assisted reading.
The original title: HUPO 蛋白质组学标准化项目 | 数据标准化挑战(2)

1固有无序蛋白 (Intrinsically Disordered Proteins, IDPs)
固有无序蛋白/区域(Intrinsically disordered proteins/region)指的是那些在正常生理状态下内在无序、没有稳定三维结构的蛋白质/区域。IDPs/IDRs广泛参与转录翻译调节、小分子储存、细胞信号传导、蛋白质磷酸化及疾病发生发展等重要的生理病理过程。
IDPs/IDRs的研究仍处于萌芽阶段,研究人员的当前任务是制定标准以提高实验分析结果的可重复性、注释深度及传播广泛性。因此,他们需要在相关数据大量涌现前(1)开发标准格式以储存它们的关键性结构功能信息,以及(2)开发用于描述、注释IDPs/IDRs结构功能的工具及格式。
2018年,MIADE(Minimum Information About Disorder Experiments)初稿在“固有无序蛋白质数据整合和标准化(Integration and standardisation of intrinsically disordered protein data)”会议上提出,初步定义了解读IDPs/IDRs实验结果的标准。此外,HUPO PSI固有无序蛋白工作组也在诞生于这个会议,并致力于继续优化MIADE及定义可用于描述一个实验的最少信息规范。该定义的明确将有利于 (1) 确定数据注释、存储和转化共享标准;(2)将IDPs/IDRs数据整合至ELIXIR核心数据资源库;(3)为IDPs/IDRs相关软件确定开发基准。
为实现以上目标,固有无序蛋白工作组与分子相互作用工作组合作开发了PSI-IDP XML/JSON和PSI-ID TAB格式。前者以PSI-MI XML模式为基础,能够明确呈现实验设置和蛋白质区域结构状态的分析结果,并可应用于大部分研究中。同样地,后者亦用于描述蛋白质区域的结构状态,该格式的开发遵循MIADE,便于使用者直接解读,且可应用于常用电子表格程序中。
除此以外,PSI-DI工作组还将开发PSI-ID术语表以对IDP实验流程进行描述,从而获得序列水平上的结构推断(Sequence-level structural inferences),具体包括实验方法、IDR结构术语、结构类型及状态的功能性术语、数据交换及交换机制术语等。
2质量控制 (Quality Control, QC)
随着基于质谱的蛋白质组学和代谢组学研究在过去几年的飞速发展,实验人员愈发重视质量控制(Quality control,QC)。在基于质谱技术的研究中,实验结果会因任意变量而产生极大的不稳定性(如样本处理时受到污染)。另一方面,提高实验的可重复性已然成为生物质谱实验研究中的优先事项,因此建立标准化质量管理体系以保证检测方法的准确性和稳定性尤为重要。
质量控制不仅可以量化实验中的变量,还可以为后续的数据分析提供参考,并反馈到质谱设置优化工作中。在过去10年间,关于蛋白质质谱实验的QC工具被大量开发。但这些工具大多仅应用于特定、非标准的实验及软件环境,其普遍适用性被大大限制。此外,由于每个工具提取的质谱数据类型,其用于存储、实现可视化、数据解释和交流的框架并不一致,数据间的互操作性和可比较性无法实现,这亦限制了生物质谱的应用潜力。
因此,HUPO-PSI在2016年成立了质量控制工作组,并致力于开发标准化质控数据框架——标准化质谱实验术语、数据和相关分析结果。
基于不同的质谱工作流程及其质量控制方法间存在较大差异,质量控制工作组希望通过提供基础技术支持来促进质谱实验的质量控制,而非明确地定义质控指标解析等工作。如下图所示,质量控制工作组制定了一种简单而通用的文件格式——mzQC(前身为qcML格式)为质量控制、可视化工作等内容提供数据交换支持,这也是各种高级任务的协调中心。值得一提的是,该文件格式将支持任意类型的质控指标,以适应与各种实验配置相关的质量控制信息。

▲qcML格式工作流程:用于存档、传输、分析和可视化质谱数据3
与之前的HUPO-PSI标准不同,mzQC采用的是已被广泛应用的轻量级资料交换格式JavaScript Object Notation(JSON),而非基于XML的文件格式(qcML采用的是XML)。这是因为XML文件相当冗长,而质谱文件的数据量本就相当可观,因此选用文件内存占用小的JSON更为合适,且许多编程语言都对其设有内置支持。换而言之,JSON降低了mzQC格式的使用门槛,并可通过扩展应用范围增加其影响力。
与此同时,PSI-MS术语表中的质量控制部分亦应用于补充mzQC格式,如相关质量指标的正式定义。这也使得该格式成为了可提供数据注释、存储功能和使质控数据可被重复性应用的强大机制。
3蛋白质修饰 (Protein Modification, MOD)
蛋白质由氨基酸按一定顺序排列结合形成的多肽链组成。1972年,国际理论与应用化学学会(International Union of Pure and Applied Chemistry, IUPAC)提出以氨基酸代码(如G代表Glycine)、并按照从氨基末端(N-)端到羧基末端(C-)端的顺序来命名蛋白质与多肽序列。但该命名法只能表示蛋白质的未被修饰形态,蛋白质的修饰命名法在当时并未明确。
未统一的蛋白质修饰命名法不仅导致科学出版物的数据信息混乱,还对开发蛋白质组学数据软件和资源库的工作人员造成极大困扰——难以决定使用什么符号格式来输入和输出数据,这将进一步对多肽和蛋白质数据的可访问性、相互操作性及可重用性造成极大影响。
在蛋白修饰工作组进行针对性标准规划前,蛋白质组学信息学和分子相互作用工作组就已经在制定数据交换标准(该标准的基础是标准数据交换文件格式——XML格式与Open Biomedical Ontologies(OBO)文件格式中的分级词汇表)。因此,为避免重复劳动和引入更多相互冲突的术语,蛋白修饰工作组的关键任务是确定标准术语表,以对蛋白质修饰类别进行分层级管理并对模糊及不完整的实验结果进行精确注释,这将有利于揭示编码修饰蛋白清晰完整的层次关系结构。
蛋白修饰工作组的最初任务是将三个源数据库——RESID数据库 (汇编了UniProt数据库中收录的自然发生的蛋白质翻译后修饰)、UNIMOD(自然发生的蛋白质翻译后修饰)和DeltaMass(按质量差异对蛋白修饰和质谱分解产物排序)进行系统性整理,以建立标准化术语条目。
逐渐地,蛋白修饰工作组开发了一个由分层术语表组成的标准分类法,分层术语表由蛋白质翻译后修饰术语和定义组成,通过特定关系进行逻辑连接。
2008年,蛋白修饰工作组不仅已成功整理构建了约1300个术语(包括彼时蛋白质翻译后修饰源数据库中的所有条目——439个术语源于RESID,335个术语源于UniMod,353个术语来源于Delta Mass,以及约有250个新术语被创建),还额外开发了适用于学术、商业数据库及搜索引擎的描述性标签、构建了蛋白质翻译后修饰的简称清单(简称将作为相应术语的同义词囊括在分类法中)、以及梳理了该命名法的拟议规则和建议。然而,继2014年的HUPO-PSI审查工作后,蛋白修饰工作组的工作似乎已经停滞,亦不再活跃。

4蛋白质分离 (Protein Separation, PS)
为更好地推动蛋白质鉴定工作,科研人员需要清晰地知道实验中所采取的分离技术、质谱仪设备、蛋白质鉴定工具和数据库等相关信息。除此以外,由于实验方法逐渐多样化,数据的复杂性及“破译”难度也在不断递增,为简化跨实验数据比较、分析、理解,或从数据集中得出统计性信息等工作,科研学者需要提供充分的实验背景信息。因为不充分的实验(及数据)描述可能会隐匿实验设计中的随机性或系统性错误。
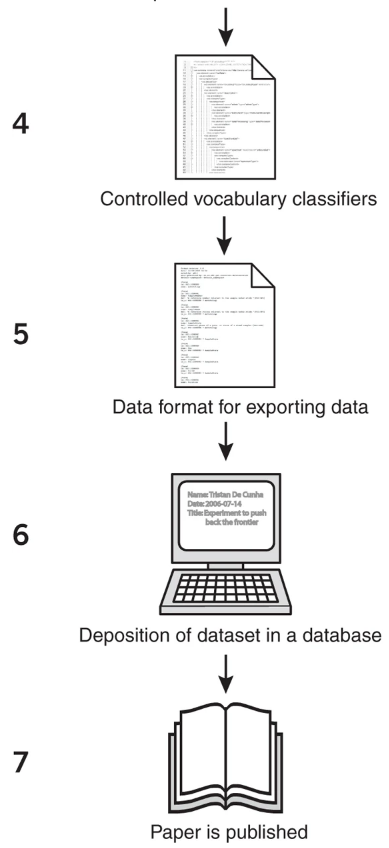
因此,研究人员建议在进行蛋白质组学数据分析时标注“背景化元数据”,以明确样品的来源和分析方式。HUPO-PSI更为此开设了凝胶电泳和质谱等蛋白质分析技术的应用指导模块,并在2007年发布了《蛋白质组学实验最少信息指南(The minimum information about a proteomics experiment, MIAPE)》。

MIAPE旨在明确研究人员向数据库提交数据集的步骤、信息等,以帮助他们顺利发表文章及相关出版物。该指南列出蛋白质组学实验中需要提供的(1)分析技术的相关信息条目,以及(2)数据分析中所需信息。
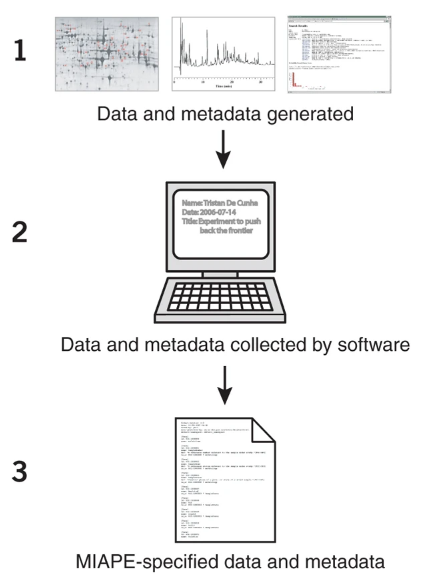
▲以MIAPE为标准的数据管理实例5
其中,蛋白质分离工作组为进一步支持MIAPE子项目中的凝胶电泳技术指导(MIAPE Gel Electrophoresis, MIAPE-GE) 工作,开发了凝胶电泳标记语言(Gel electrophoresis Markup Language,GelML)以实现凝胶数据标准化。
 ▲MIAPE-GE 指南的主要内容5
▲MIAPE-GE 指南的主要内容5
GelML融合了彼时基于凝胶的蛋白质组数据格式优点,包括AGML、HUP-ML和PEDRo。首先,AGML以二维凝胶电泳数据为中心,用自由文本元素表示样品、设备和蛋白质检测程序,并包含用于表示凝胶图像特征强度的组件。HUP-ML也以二维凝胶电泳数据为中心,可记录电泳中使用的溶液及跑胶时长。值得一提的是,HUP-ML开发于2002年(HUPO-PSI成立前),在早期的HUPO-PSI会议上被视作凝胶建模的起点。而PEDRo模型则旨在描述蛋白质组学实验中的数据流,且能够捕获到关于凝胶技术的部分重要信息。但PEDRo无法对凝胶图像采集流程和结果图像进行充分注释,亦无法存储电泳技术运行条件的相关信息。
基于以上信息以及PEDRo和AGML研发人员的加入,HUPO-PSI于2005年举行的PSI秋季会议上开始取长补短的建模工作,并于2007年底发布GelML 1.0.0版本,得到学术界、商业界同行的广泛支持。2010年,研究人员通过将建模语言从UML调整为XML格式,将开发方法改为GelML XSD和FuGElight XSD,并将GelML 1.0.0升级为GelML 1.1.0版本。相关文章亦记录,与PSI-MOD工作组一样,PSI-PS工作组也在2014年PSI会议后不再活跃 。

▲GelML格式的处理流程,以及每个部分都将捕获某些关键细节6
5总结
蛋白质中蕴藏着无数把解开生命奥秘的关键钥匙。蛋白质组学发展至今才短短20余年,生命科学等相关领域的科研人员和临床医生在其间奋力推动蛋白质组学向更广阔的研究领域挺进,突破数据信息统一标准,以促进数据交流共享的瓶颈显得愈发关键。HUPO-PSI所设立的这7个子项目亦不负众望在人类大健康命题中奋楫中流,历年来的临床转化、上游科研突破都包裹着它们的身影。同时,我们亦希望更多的研究人员能加入HUPO,促进上游科研创新,下游商业转化,为人类大健康命题出一分力!
参考资料
1. Liu, Y., Chen, S., Wang, X., & Liu, B. (2019). Identification of intrinsically disordered proteins and regions by length-dependent predictors based on conditional random fields. Molecular Therapy-Nucleic Acids, 17, 396-404.
2. Davey, N. E., Babu, M. M., Blackledge, M., Bridge, A., Capella-Gutierrez, S., Dosztanyi, Z., … & Tosatto, S. C. (2019). An intrinsically disordered proteins community for ELIXIR. F1000Research, 8.
3. Bittremieux, W., Walzer, M., Tenzer, S., Zhu, W., Salek, R. M., Eisenacher, M., & Tabb, D. L. (2017). The Human Proteome Organization–Proteomics Standards Initiative Quality Control Working Group: Making Quality Control More Accessible for Biological Mass Spectrometry. Analytical chemistry, 89(8), 4474-4479.
4. Montecchi-Palazzi, L., Beavis, R., Binz, P. A., Chalkley, R. J., Cottrell, J., Creasy, D., … & Garavelli, J. S. (2008). The PSI-MOD community standard for representation of protein modification data. Nature biotechnology, 26(8), 864-866.
5. Gibson, F., Anderson, L., Babnigg, G., Baker, M., Berth, M., Binz, P. A., … & Jones, A. R. (2008). Guidelines for reporting the use of gel electrophoresis in proteomics. Nature biotechnology, 26(8), 863-864.
6. Gibson, F., Hoogland, C., Martinez‐Bartolomé, S., Medina‐Aunon, J. A., Albar, J. P., Babnigg, G., … & Jones, A. R. (2010). The gel electrophoresis markup language (GelML) from the Proteomics Standards Initiative. Proteomics, 10(17), 3073-3081.